
分子模拟的首要目标是准确预测分子体系的实验值,其长远目标之一为设计适用于任意中性有机分子且几乎不依赖于实验数据的计算模型。自由能是生物物理系统最重要、同时也最难以预测的物理量。其中,溶剂化自由能的计算是描述生物物理过程的基础。
模拟软件包可通过区域分子力场计算自由能,通常通过与经验观测值的拟合得出部分或全部参数,存在两个缺点。首先,现有的实验数据不足以准确描述现有化合物的模型;其次,因难以确定错误来源,经验模型的错误很难被纠正。相比之下,基于量子力学(QM)的参数化分子模型可以克服这些问题。量子力学可计算任意分子,不依赖于实验数据,且可通过对相互作用能量的非精确描述追溯预测误差,并在模型中进行纠正。
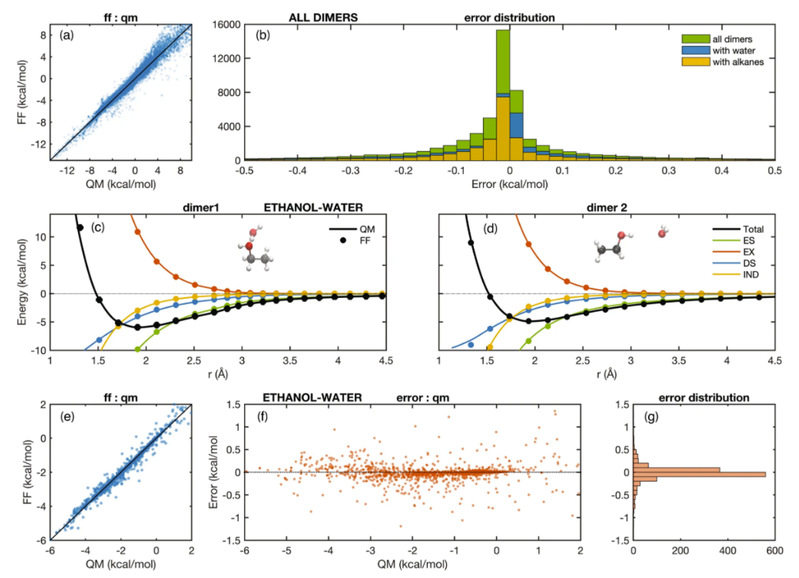
最近,Michael Levitt教授和合作者共同开发了ARROW FF,这是一个极化力场模型,基于第一性原理的量子力学计算来预测各种中性有机化合物的溶解自由能。通过模拟QM参数化力场,模型可涵盖任意有机分子,并预测分子系统的溶解自由能,对中性溶液来说,其预测精度可达约0.3千卡/摩尔。作者们通过计算水、环己烷中多种化学功能团的溶解自由能来证明该模型及模拟机制的预测能力。
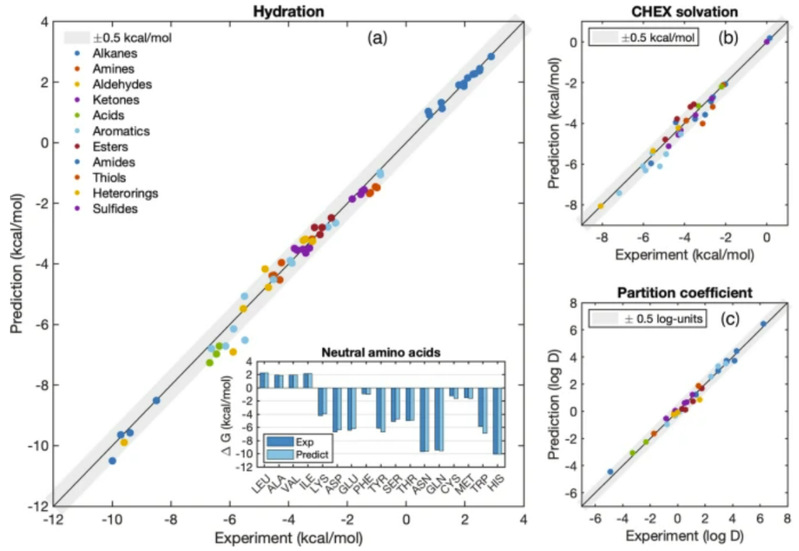
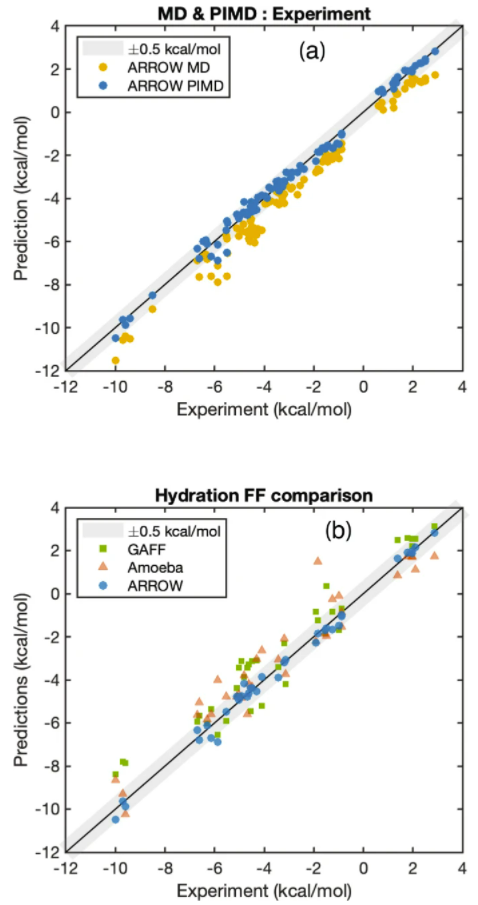
ARROW FF模型具有多极性、可极化性,其计算效率与目前最先进的同类模型相当。值得注意的是,核量子效应(NQE)在自由能计算中起着重要作用,通过适当考虑核运动的量子性质,可使预测结果系统地靠近实验值,并可将MAE的预测误差从0.78(黄点)降低到0.2(蓝点)千卡/摩尔。与两个广泛使用的经验模型GAFF和AMOEBA(前者是许多固定电荷模型的代表,后者是一个可极化模型)相比,ARROW FF的预测精度是两者的三倍以上。
本研究基于第一性原理,构建了可广泛使用的分子模型ARROW FF,该模型可提高中性有机化合物的溶解自由能的预测精度,代表生物分子模拟的重要进展。
研究发表于Nature Communications,阅读全文请访问 https://www.nature.com/articles/s41467-022-28041-0,代码、工具包请参见Github https://github.com/freecurve/interx_solvation_suite.
今日编辑:徐 敏
