
The primary goal of molecular simulations is to accurately predict experimental observations of molecular systems. In the long term, one of their goals is also to develop computational models that are applicable to arbitrary neutral organic molecules and are virtually independent of experimental data. The most important parameter for biophysical synthesis is the free energy, and it is also the most difficult one to predict.
Modern, areal molecular force fields in simulation packages that allow free energy calculations derive some or all of their parameters by fitting them to empirical observations, but with two drawbacks. First, available experimental data are insufficient to produce models that accurately describe existing compounds, and second, error correction of empirical models is difficult because it is hard to identify the source of the error. In contrast, Quantum Mechanical (QM)-parametrized physics-based molecular models can overcome these two issues. Quantum mechanical calculations do not depend on experimental data and could be obtained for any arbitrary moleculesand, and prediction errors can be traced to the imprecise description of the interaction energies and rectified in the model.
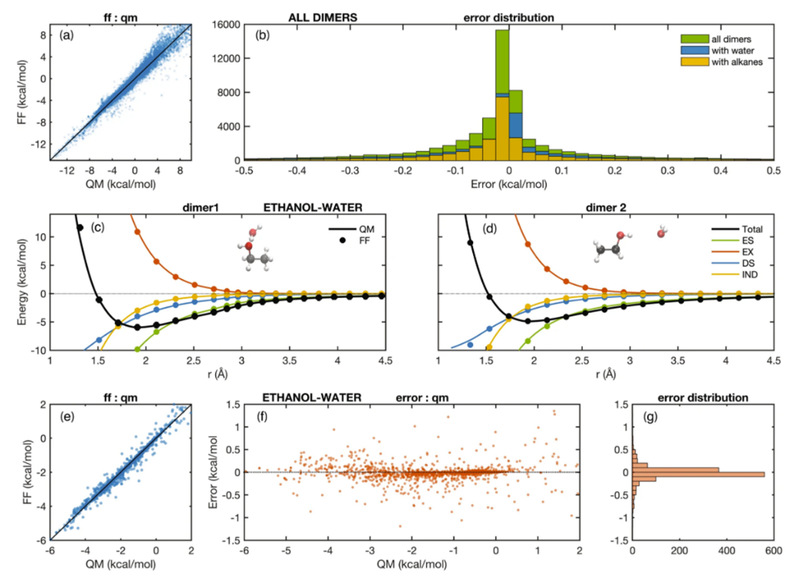
In this work, Professor Michael Levitt and co-authors developed a polarization force field model, ARROW FF to predict the free energy of solvation of various neutral organic compounds based on quantum mechanical calculations of the first-nature principle. They implemented a QM-parametrized force field in a simulation stack that covers arbitrary organic molecules and predicts solvation free energies of molecular systems to accuracy of ~0.3kcal/mol for neutral species, and they demonstrate the predictive ability of the model and simulation machinery by computing solvation free energies for a wide range of chemical functional groups in water and cyclohexane.
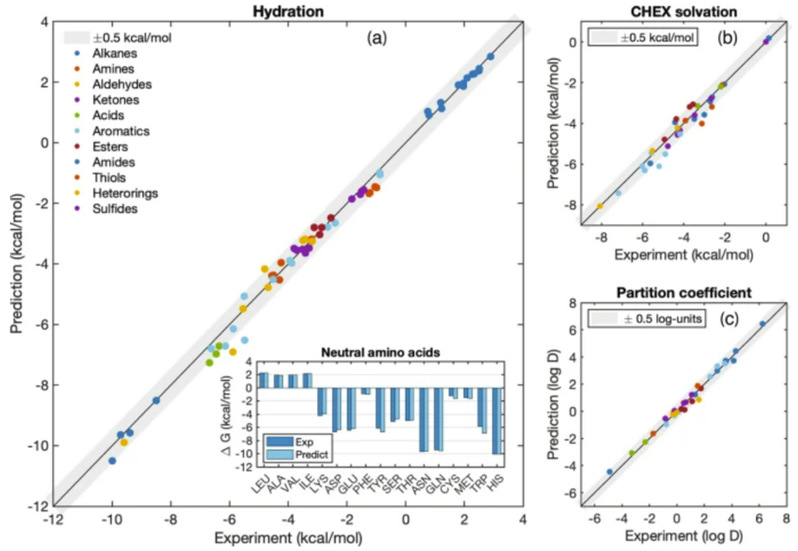
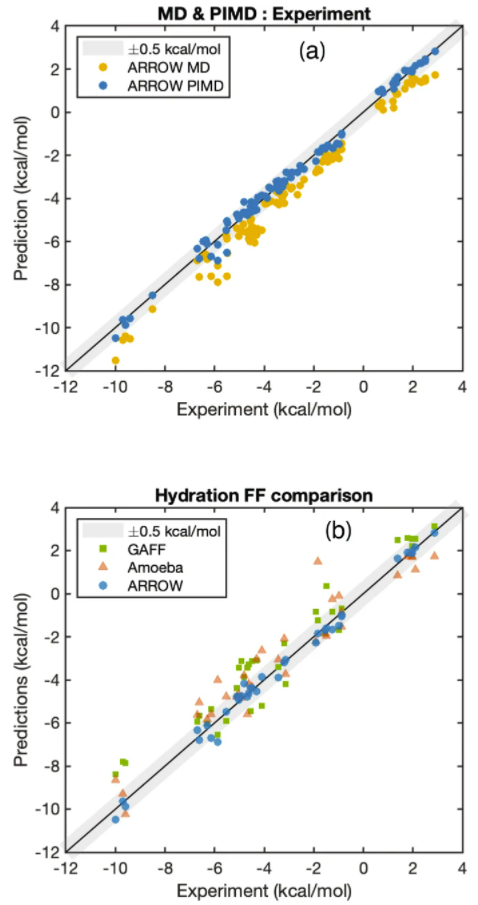
The ARROW FF model is multipolar and polarizable, and its computational efficiency is comparable to the most advanced software of its kind currently available. It is notable that nuclear quantum effects (NQE) plays an important role in the free energy calculation and proper accounting of the quantum nature of nuclear motion systematically shifts the predictions toward the experimental values and improves the prediction error of MAE from 0.78 (yellow dot) to 0.2 (blue) kcal/mol. To compare with two widely used empirical models GAFF and AMOEBA (the former is a representative of many fixed-charge models, the latter is a polarizable model), the prediction accuracy of ARROW FF is more than three times that of the two.
This work represents a major advance in biomolecular simulation by constructing a wide-coverage molecular modelling toolset from first principles with excellent predictive ability in the liquid phase.
The work is published by Nature Communications at https://www.nature.com/articles/s41467-022-28041-0 and the codes, tools and data needed to reproduce the data presented in this article is available on github https://github.com/freecurve/interx_solvation_suite.
About Professor Michael Levitt
Dr Michael Levitt is a Professor, by Courtesy, of Computer Science at Department of Structural Biology,Stanford University. He is the Laureate for The Nobel Prize in Chemistry 2013, for the development of multiscale models for complex chemical systems.. Dr Michael Levitt is an adjunct professor of Shanghai Institute for Advanced Study of Zhejiang University (SIAS).
Here is how he described his work and the research from his lab at Stanford University:
“I pioneered of computational biology setting up the conceptual and theoretical framework for a field that I am still actively involved in at all levels. More specifically, I still write and maintain computer programs of all types including large simulation packages and molecular graphics interfaces. I have also developed a high-level of expertise in Perlscripting, as well as in the advanced use of the Office Suite ofprograms (Word, Excel and PowerPoint), which is more important and rare than it may seem. My research focuses on three different but inter-related areas of research. First, we are interested in predicting the folding of a polypeptide chain into a protein with a unique native-structure with particular emphasis on how the hydrophobic forces affect the pathway. We expect hydro phobic interactions to energetically favor structure that are more native-like. In this way, the same stabilizing interactions that exist in the final folded state the search tractable. Second we are interested in predicting protein structure from sequence without regard for the process of folding. Such prediction relies on the well-established paradigms that similar protein sequences imply similar three-dimensional structures. We have focused on the hardest problem in homology modeling: the refinement of a near-nativestructure to make it more precisely like the actual native structure of protein. We have also focused on how the general similarity of all protein sequences resulting from their evolution from common ancestorsequence affects the nature of the protein universe. Third, we are focusing on meso scale modeling of large macromolecular complexes suchas RNA polymerase and the mammalian chaperonin. In this work, done in close collaboration with experimentalists, we use new morphingstrategies combined with normal mode analysis in torsion angle spaceto overcome problems caused by the size and complexity of thesecritical, biomedically important systems. All this work depends on the way a molecular structure is represented in terms of the force-field that allows calculation of the potential energy of thesystem. We employ a very wide variety of such energy functions that extend from knowledge-based statistical potentials for a single interaction center per residue to quantum-mechanical force-fields that include inductive effects as well as polarization.”
About SIAS
Shanghai Institute for Advanced Study of Zhejiang University (SIAS) is a jointly launched new institution of research and development by Shanghai Municipal Government and Zhejiang University in June, 2020. The platform represents an intersection of technology and economic development, serving as a market leading trail blazer to cultivate a novel community for innovation amongst enterprises.
SIAS is seeking top talents working on the frontiers of computational sciences who can envision and actualize a research program that will bring out new solutions to areas include, but not limited to, Artificial Intelligence, Computational Biology, Computational Engineering and Fintech.