
The usual method for predicting the structure of a protein using artificial intelligence is to model the main chain of the protein and then model the side chains based on the conformation of the main chain. Proteins usually contain 20 amino acids with nearly identical main chains and very different side chains. Since the vast majority of sites where drug molecules bind to human proteins are on the side chains, accurate side chain prediction by artificial intelligence (AI) techniques can be of great value in the development of new drugs. Such prediction capability can also be used to explain the mechanisms of point mutations and small fragment mutations in genes, providing valuable insights for the study and treatment of genetic diseases.
Accurate modelling of protein side chains is essential for protein folding and design. In recent studies, researchers have developed algorithms for modelling side chains, most of which are based on random sampling, such as SCWRL4 and OPUS -Rota3, by selecting from a discrete library of side-chain dihedral rotors and then optimizing them via a series of energy functions to find the dihedral rotor with the lowest energy as the final result. The advantage of the sampling-based side-chain modelling algorithm is that it is faster, but its overall accuracy in side-chain prediction needs to be improved because it uses discrete rotors and is limited by the accuracy of the energy function.
Over the years, Professor Jianpeng Ma of Fudan University has led a team that has developed a series of OPUS algorithms to predict the 3D structure of protein main and side chains using AI techniques. A recent paper published by his team shows that OPUS -Rota4 can predict side chain structure 13% better than AlphaFold2, based on the main chain structure of several proteins predicted by AlphaFold2 in an international protein structure prediction competition.
The introduction of deep learning algorithms in OPUS -Rota4 has led to a significant improvement in the accuracy of protein side chain modelling. In this paper, the authors present a set of open-source tools for protein side chain modelling consisting of three modules: OPUS -RotaNN2, for predicting the dihedral angles of protein side chains; OPUS -RotaCM, for measuring the distance and direction between side chains of different residues; and OPUS -Fold2, a modelling framework that uses the information derived from the above two modules for side chain modelling.
The team first used OPUS -RotaNN2 to obtain the initial side-chain dhedral angle predictions by combining various extracted features, then OPUS -RotaCM was employed to obtain the side-chain atom contact maps, and finally OPUS -Fold2 was used to optimise the initial side-chain dhedral angle predictions based on the contact maps and output the final results.
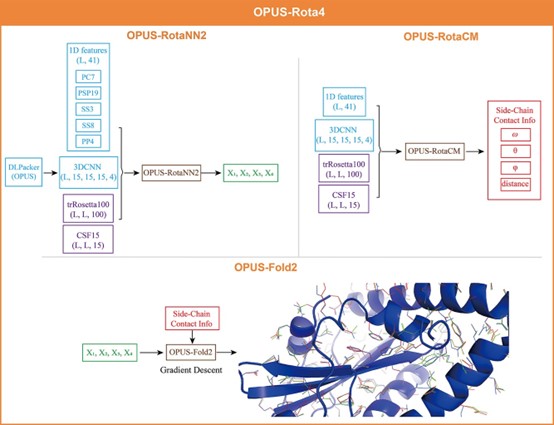
OPUS -Rota4 framework
Authors performed tests using three natural conformational test sets: CAEMO (60) with 60 test proteins, CASPFM (56) with 56 test proteins, and CASP14 (15) with 15 test proteins. Their results showed that OPUS -Rota4 outperformed other side-chain modelling algorithms in all three test sets.
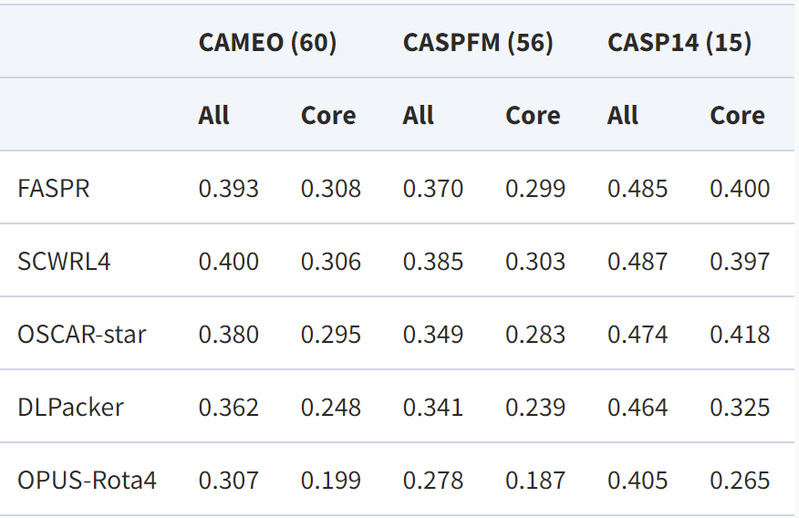
RMSD results for the three natural conformation test sets: lower values indicate closer to the natural conformation, All for all residues and core for central residues. All residues contain both central and surface residues. The central residues are located inside the protein and are more important for its biological function.
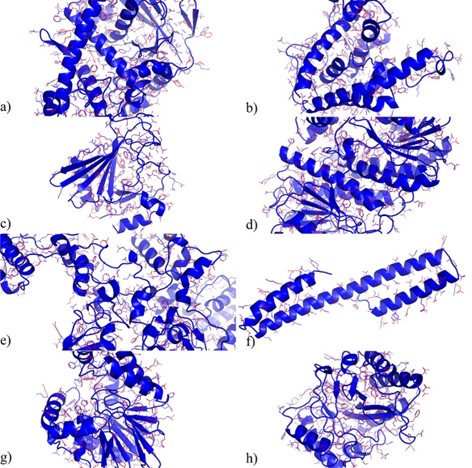
Predicted structures of 15 proteins in CASP14 (15)
The results presented in the paper showed that the side-chain predictions of OPUS-Rota4 are generally close to the natural conformation, especially for the central residues located inside the protein, where the predictions strongly overlap with the natural conformation.
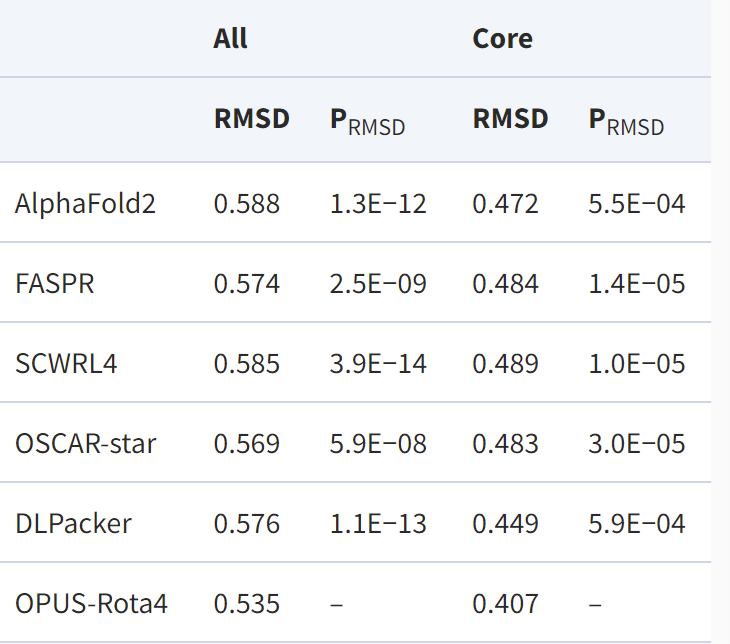
In addition to the three sets of tests for the natural conformation, the authors used AlphaFold2 to obtain the predicted structures for 15 proteins in CASP14 (15) and re-modelled their side chains in various ways based on the predicted backbone structures. The results showed that OPUS -Rota4 gave significantly better results than other methods for modelling side chains and that the side chains were closer to the natural conformation than those predicted by AlphaFold2.
The team also analyzed several relatively poorly predicted structures. Results suggested that the main reason for the poor predictions may be the presence of a long disordered loop region in each of these structures, which has a high degree of structural freedom in the amino acid side chain. Further research on protein side-chain modelling will be conducted to improve accuracy and investigate the application of side-chain modelling to practical problems.
The use of AI to accurately predict protein side chain structures is not only for life sciences, but also a breakthrough for computational biology., said Professor Ma.
To read the full article, please visit https://academic.oup.com/bib/article/23/1/bbab529/6461160.
With courtesy to Shanghai Artificial Intelligence Laboratory.
About Professor. Jianpeng Ma
Dr Ma is a Fellow of the American Society for Medical Bioengineering, an Elected Fellow of the American Association for the Advancement of Science(AAAS), an Elected Fellow of the American Physical Society(APS), and a recipient of the 2004 Norman Hackermann Award for Chemical Research.
He joined Fudan University, China in 2018 after being Lodwick T. Bolin Professor in Biochemistry for Baylor College of Medicine and Rice University. Together with Professor Michael Levitt, he founded The Multiscale Research Institute for Complex Systems(MRICS) at Fudan University and served as its Dean.
Dr Ma’s research interests include biophysics, computational biology, and structural biology. He is dedicated to the development of new computational methods for the study of biological systems that can be used to overcome the difficulties in experimental research and to solve important problems in complex biological systems in combination with experimental methods.
About SIAS
Shanghai Institute for Advanced Study of Zhejiang University (SIAS) is a jointly launched new institution of research and development by Shanghai Municipal Government and Zhejiang University in June, 2020. The platform represents an intersection of technology and economic development, serving as a market leading trail blazer to cultivate a novel community for innovation amongst enterprises.
SIAS is seeking top talents working on the frontiers of computational sciences who can envision and actualize a research program that will bring out new solutions to areas include, but not limited to, Artificial Intelligence, Computational Biology, Computational Engineering and Fintech.