
Recently, Jianpeng Ma, Adjunct Professor of Computing + Biology Lab at Shanghai Institute for Advanced Studies, Zhejiang University (SIAS), Director of the Multiscale Research Institute for Complex Systems,Fudan University, and his team proposed OPUS-Mut protein side-chain modeling algorithm, demonstrating its potential in application to protein interaction problems while further improving the accuracy of side-chain modeling. The results are now accessible at http://github.com/OPUS-MaLab/opus_mut and the article Studying protein-protein interaction through side-chain modeling method OPUS -Mut was published online in Briefings in Bioinformatics. These results provide a powerful tool for new drug development.
The three-dimensional structure of a protein consist of a main chain and a side chain. In nature, proteins contain 20 amino acids with almost identical main chains and very different side chains. Since the vast majority of sites where drug molecules bind to human proteins are on the side chains of the amino acids, the ability to accurately predict the positions of side chains is of great importance for the development of new drugs. It could also be used to explain the mechanism of point mutations and small fragment mutations in genes, providing valuable ideas for the study and treatment of genetic diseases.
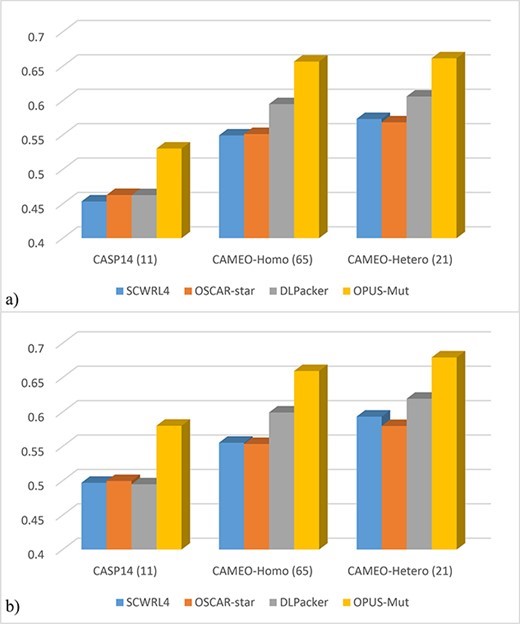
The side-chain structure of proteins is the basis for protein-protein interactions, which in turn are key to peptide drug development. When proteins interact with each other, the side-chain structure of their residues located at the contact surface plays a key role and undergoes a corresponding conformational change. Accurate modeling of the side chains of interfacial residues is therefore critical for the study of protein interactions.
In modeling the side chains of protein multimers, especially for amino acid side chains located at the contact surface, OPUS-Mut shows higher accuracy compared to SCWRL4, OSCAR-star and DLPacker on three oligomer datasets CASP14 (11), CAMEO-Homo (65) and CAMEO-Hetero (21). More accurate prediction of amino acid side chain conformations for interfacial residues could support the study of protein interaction problems.
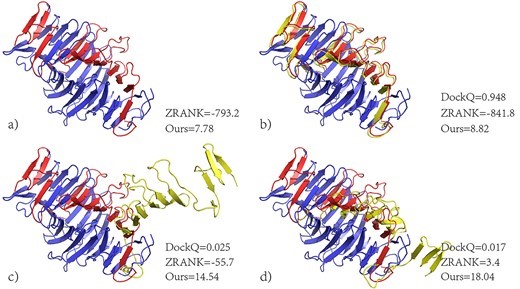
In protein docking conformation scoring, accurate prediction of side-chain conformations is key to improving the accuracy of scoring functions based on side-chain position information. OPUS-Mut indicates the confidence level of the predicted side chain along with the predicted side chain of the residue. This confidence level can also indicate whether the environment of the residue is comfortable or not. The higher the confidence level, the easier it is to place the side chain of the residue in its environment, which is closer to the natural conformation of the training set.
This method of assessing the docking conformation by feedback of the side chain to the local environment highlights the importance of side chain in docking and is a good complement to many other current assessment functions. This facilitates the screening of protein docking conformations and the modeling of their interaction processes, which is critical for peptide drug development.
The results show that OPUS-Mut based scoring function is superior to scoring functions such as ZRANK, GNN-DOVE in distinguishing natural conformations from virtual docking conformation sets.
The article can be accessed at Studying protein–protein interaction through side-chain modeling method OPUS-Mut | Briefings in Bioinformatics | Oxford Academic (oup.com) and the code and pre-trained models of OPUS-Mut and the four datasets used in this paper can be found at http://github.com/OPUS-MaLab/opus_mut, which are free to academic usage only.
Jianpeng Ma's team has been devoted to modeling protein side chain and has developed a series of algorithms such as OPUS-Rota, OPUS-Rota2, OPUS-Rota3, OPUS-Rota4, and OPUS-Mut, corresponding to various application scenarios and to support peptide drug development.
About Professor Jianpeng Ma
Dr. Ma is a Fellow of the American Society for Medical Bioengineering, an Elected Fellow of the American Association for the Advancement of Science(AAAS), an Elected Fellow of the American Physical Society(APS), and a recipient of the 2004 Norman Hackermann Award for Chemical Research.
He joined Fudan University, China in 2018 after being Lodwick T. Bolin Professor in Biochemistry for Baylor College of Medicine and Rice University. Together with Professor Michael Levitt, he founded The Multiscale Research Institute for Complex Systems(MRICS) at Fudan University and served as its Dean.
Dr Ma’s research interests include biophysics, computational biology, and structural biology. He is dedicated to the development of new computational methods for the study of biological systems that can be used to overcome the difficulties in experimental research and to solve important problems in complex biological systems in combination with experimental methods.
About SIAS
Shanghai Institute for Advanced Study of Zhejiang University (SIAS) is a jointly launched new institution of research and development by Shanghai Municipal Government and Zhejiang University in June, 2020. The platform represents an intersection of technology and economic development, serving as a market leading trail blazer to cultivate a novel community for innovation amongst enterprises.
SIAS is seeking top talents working on the frontiers of computational sciences who can envision and actualize a research program that will bring out new solutions to areas include, but not limited to, Artificial Intelligence, Computational Biology, Computational Engineering and Fintech.