
Molecular dynamics (MD) has evolved into a ubiquitous, versatile and powerful computational method for fundamental research in science branches such as biology, chemistry, biomedicine and physics over the past 60 years. Powered by rapidly advanced supercomputing technologies in recent decades, MD has entered the engineering domain as a first-principle predictive method for material properties, physicochemical processes, and even as a design tool.
In light of this, Professor Kaihong Luo from Shanghai Institute for Advanced Study, Zhejiang University illustrated such developments and their far-reaching consequences, with a focus on MD for combustion and energy systems encompassing topics like gas/liquid/solid fuel oxidation, pyrolysis, catalytic combustion, heterogeneous combustion, electrochemistry, nanoparticle synthesis, heat transfer, phase change, and fluid mechanics.
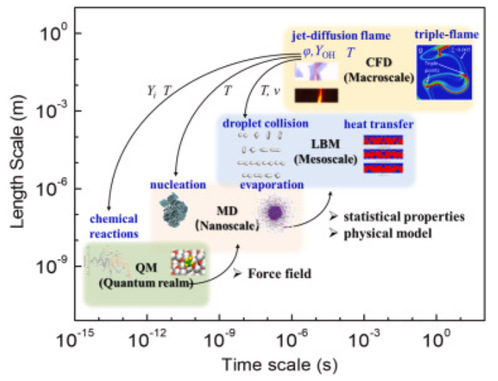
Overview of the time and length scales for multiscale modeling in combustion
First, the authors systemically described the theoretical framework of the MD methodology, covering both classical and reactive MD. The emphasis was on the development of the reactive force field (ReaxFF) MD, which enables chemical reactions to be simulated within the MD framework, utilizing quantum chemistry calculations and/or experimental data for the force field training.
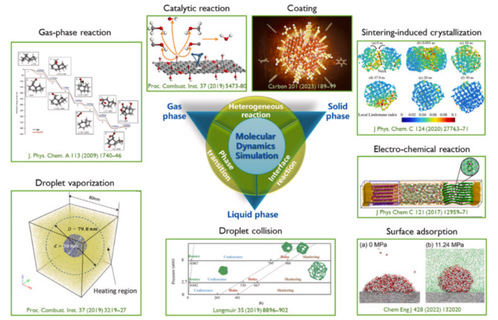
Schematic of selected combustion and energy topics studied with the MD method, showing phase transitions and interplays among gas, liquid, and solid phases
The authors then provided details of the numerical methods, boundary conditions, post-processing and computational costs of MD simulations.

Relations among different MD methods with quantum mechanics(QM), semi-empirical method, and molecular mechanics(MM)
This was followed by a critical review of selected applications of classical and reactive MD methods in combustion and energy systems. They laid out the simulation procedure, and listed software and visualization tools for MD simulation and postprocessing. The authors further demonstrated that the ReaxFF MD has been successfully deployed to gain fundamental insights into pyrolysis and/or oxidation of gas/liquid/solid fuels, revealing detailed energy changes and chemical pathways.
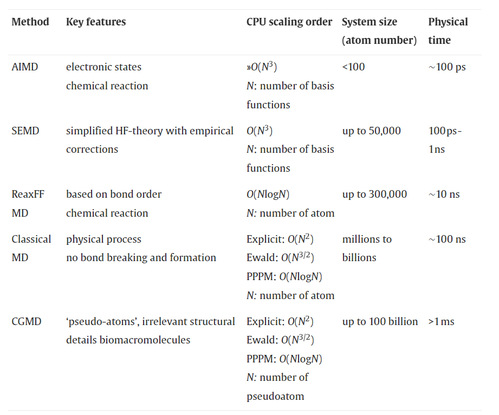
Comparison among MD methods
The authors continued to explain the complex physico-chemical dynamic processes in catalytic reactions, soot formation, and flame synthesis of nanoparticles from an atomistic perspective. Flow, heat transfer and phase change phenomena are also scrutinized by MD simulations. Unprecedented details of nanoscale processes such as droplet collision, fuel droplet evaporation, and CO2 capture and storage under subcritical and supercritical conditions are examined at the atomic level.
The authors concluded that MD methods are most suited to nano-sized systems with concentrated atoms/molecules and highly dynamic processes (i.e., short time duration). For classical MD, these include (but are not confined to): nanodroplet dynamics, flow in nanochannels, and phase change. For ReaxFF MD, examples include: nanoparticle combustion, soot formation and heterogeneous catalytic combustion. For phenomena/processes that span a large volume and/or are evolving slowly, MD simulations are not cost-effective on current computing platforms.
Classical MD has found numerous applications that are of relevance to combustion and energy systems. As stated above, typical applications involve nanodroplets, nanoparticles and nanochannels, yielding new insights. For example, MD has been used to simulate the dynamics of droplet-droplet collision, droplet-wall collision, droplet evaporation, etc. MD has revealed the dramatic change in the interfacial properties from subcritical to supercritical evaporation of droplets. Moreover, in the MD simulation of nanoparticle sintering, phase change or crystallization has been found following the merge of two amorphous nanoparticles. For flow in nanochannels, MD allows both slip and no-slip wall conditions, as the physics of flow plays out naturally, without prior assumptions. An example of advanced applications is the MD simulation of shale gas recovery in geological formations, which is enhanced by injected carbon dioxide. The simulation reveals intricate interactions among the gas species, and between the gases and the porous matrices. In one study, the hydrophobicity of the substrate is changed due to atomic interactions between water and carbon dioxide molecules. All these findings are impossible to achieve using continuum-based macroscopic simulations. Finally, MD simulations can also be extended to flow through membranes, transport of charged nanoparticles, bioflow over soft matters such as cells.
The authors further believed that ReaxFF MD had become a vital research tool, gaining unprecedented insights into the atomic and molecular behaviors of combustion and energy systems. For gaseous fuels, ReaxFF MD can reveal all the intermediate species and detailed chemical pathways in pyrolysis and combustion processes. For fuels in the form of nanodroplets and nanoparticles, ReaxFF MD can show detailed dynamics of atomic diffusion, bond-breaking, bond formation as well as energy conversion. Fruitful research based on ReaxFF MD has been conducted on initial soot formation, in which gaseous PAHs eventually become incipient solid soot particles after going through a series of physical and chemical processes. A recent extension is the ReaxFF MD of flame-synthesized nanoparticles under simplified and controlled environments. Furthermore, ReaxFF MD has been deployed to understand the mechanisms of catalytic reactions, in which the nanoparticle catalysts, the substrates, and gaseous species form multi-way interactions. Finally, both the classical and ReaxFF MD methods have been extensively employed to multi-physicochemical processes in batteries and fuel cells. It is now feasible to perform MD simulations of the time-dependent charging and discharging of a complete battery cell consisting of electrodes, electrolytes and solid electrolyte interphase layers, showing detailed transport of ions and electrons. Undoubtedly, neither QM methods nor continuum-based numerical simulations could provide the rich details of the above atomic and/or molecular behaviors over the same length and time scales.
Finally, the outlook for atomistic simulations of combustion and energy systems is discussed in the context of emerging computing platforms, machine learning and multiscale modelling.
The review was published as ‘Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems’ in Progress in Energy and Combustion Science, and could be accessed at https://www.sciencedirect.com/science/article/pii/S036012852300014X.