
Pancreatic cancer is a leading cause of cancer-related mortality globally. The incidence of pancreatic cancer has risen drastically over the past decades[1]. Surgical intervention is only feasible for 10–15% of patients with localized disease, most incidences initially necessitate neoadjuvant or palliative systemic treatment[2]. As an emerging therapeutic strategy, targeted therapy offers the potential for greater precision and effectiveness by targeting specific molecular markers with reduced systemic toxicity, thus enjoying wide application in tumor treatment[3]. Currently, overall survival benefits of targeted drugs designed for specific mutations in pancreatic cancer remain limited, and progress in the development has been hindered by small patient cohorts enrolled in these trials. Therefore, enhanced and broader therapeutic targets for pancreatic cancer need to be identified urgently.
On May 17, 2024, Professor Ruhong Zhou, Zhejiang University, along with Professors Xueli Bai and Liang Tingbo from the First Affiliated Hospital, Zhejiang University School of Medicine, published the work Inhibition of DEF-p65 Interactions as a Potential Avenue to Suppress Tumor Growth in Pancreatic Cancer in Advanced Science. The study systematically investigated the role of DEF in pancreatic cancer progression and discovered that DEF binds to p65, a key transcription factor in the NF-κB pathway, inhibiting its ubiquitination and degradation. The findings demonstrate that DEF serves as a prognostic factor in pancreatic cancer. DEF interacts with and stabilizes p65, protecting it from ubiquitination-mediated degradation, thereby promoting tumor proliferation in pancreatic cancer. Subsequently, by elucidating the underlying binding mechanism, a specific peptide-031 was successfully developed to disrupt the formation of the DEF-p65 complex. Remarkably, peptide-031 exerts promising anti-tumor effects both in pancreatic cancer cells and mouse models.
DEF is a key gene for maintaining the normal development of digestive organs such as the liver, pancreas, and small intestine in juvenile[4]. DEF is indispensable for the binding of p53 protein to Caplain3 in the nucleolus accumbens, facilitates the degradation of the p53 protein and prevent apoptosis[5]. Instead of acting as a guardian of the genome, mutant p53 can acquire oncogenic properties, thereby disrupting key cellular processes and contributing to the initiation and progression of pancreatic cancer. However, the precise role of DEF in the progression of pancreatic cancer, along with its potential interactions with pathways associated with pancreatic oncogenesis in humans, continue to be subjects of significant interest. Therefore, investigating the role of DEF in pancreatic cancer progression appears to be a promising option for revealing the pathogenesis of pancreatic cancer and developing new therapeutic targets.
Through analysis and experimental validations of public databases and clinical samples from pancreatic cancer patients at the First Affiliated Hospital, Zhejiang University School of Medicine, the authors found that DEF proteins were highly expressed in patients and were associated with poorer survival prognosis. It was observed that the depletion of DEF inhibited the NF-κB pathway without impacting the p53 pathway. DEF interacts with the p65 protein, the key subunit of the NF-κB transcription factor complex, preventing ubiquitination-induced degradation and promoting tumor cell survival.
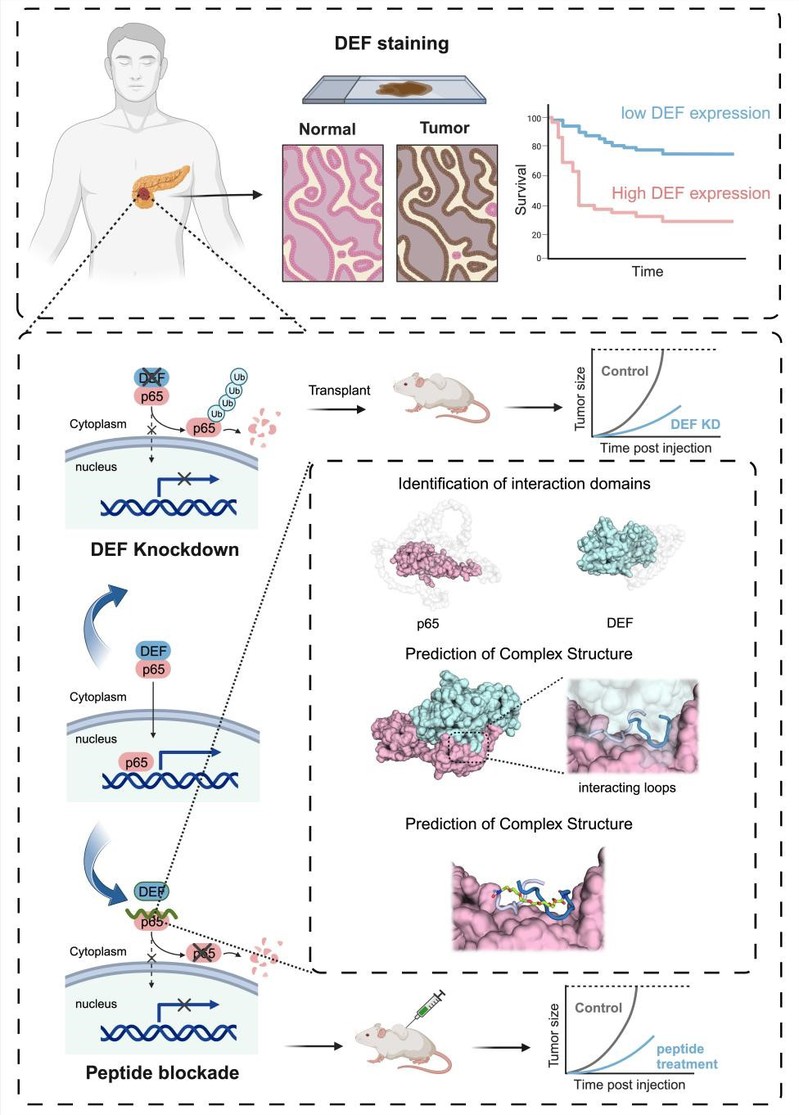
Figure 1. Schematic illustration of the design and mechanism of DEF-mimicking peptide for anti-tumor therapy in pancreatic cancer.
The authors further identified specific binding sites where DEF forms a complex with the p65 protein via computational methods. Based on these findings, they mimicked this binding mode and designed a blocking peptide (peptide-031) that targeted p65 and disrupted the critical interaction between DEF and p65. Subsequent in vitro and in vivo experiments demonstrated that treating pancreatic cancer cells with peptide-031 effectively downregulated p65 expression and inhibited tumor growth. Overall, the findings highlight the oncogenic role of DEF in pancreatic cancer while establishing it as a valuable therapeutic target for intervention.
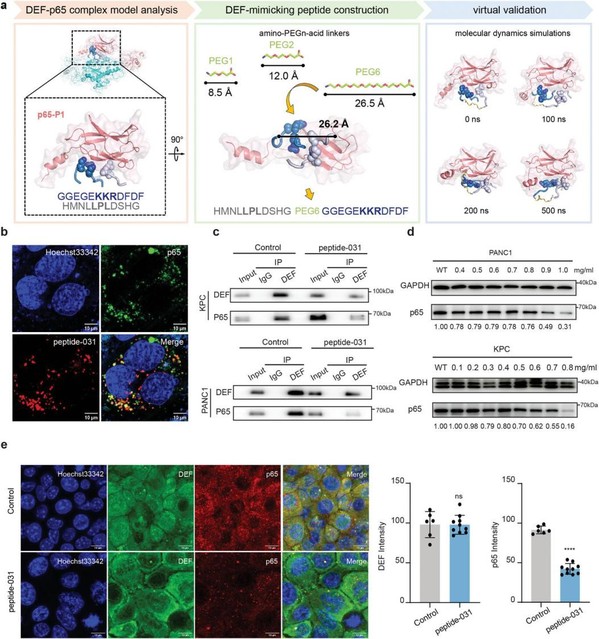
Figure 2. DEF-mimicking peptide that specifically reduces p65 expression was designed. a) Computational virtual design flow of DEF-mimic peptides targeting p65.
The work demonstrated medical-industrial-industrial joint model for tumor drug development, reflecting multidisciplinary advantages of Zhejiang University. This work was supported by the National Key Research and Development Program, National Natural Science Foundation of China, the National Independent Innovation Demonstration Zone Shanghai Zhangjiang Major Projects, the National Center of Technology Innovation for Biopharmaceuticals, and the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study. To access the article please visit https://onlinelibrary.wiley.com/doi/10.1002/advs.202401845.
Reference:
1. Morrison, A.H., Byrne, K.T., and Vonderheide, R.H. (2018). Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 4, 418-428.
2. Collisson, E.A., Sadanandam, A., Olson, P., Gibb, W.J., Truitt, M., Gu, S., Cooc, J., Weinkle, J., Kim, G.E., Jakkula, L., et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17, 500-503.
3. Halbrook, C.J., Lyssiotis, C.A., Pasca di Magliano, M., and Maitra, A. (2023). Pancreatic cancer: Advances and challenges. Cell 186, 1729-1754.
4. Chen, J., Ruan H Fau - Ng, S.M., Ng Sm Fau - Gao, C., Gao C Fau - Soo, H.M., Soo Hm Fau - Wu, W., Wu W Fau - Zhang, Z., Zhang Z Fau - Wen, Z., Wen Z Fau - Lane, D.P., Lane Dp Fau - Peng, J., and Peng, J. (2005). Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev.
5. Tao, T., Shi, H., Guan, Y., Huang, D., Chen, Y., Lane, D.P., Chen, J., and Peng, J. (2013). Def defines a conserved nucleolar pathway that leads p53 to proteasome-independent degradation. Cell Res 23, 620-634.